¿Qué son las distrofias de conos y bastones?
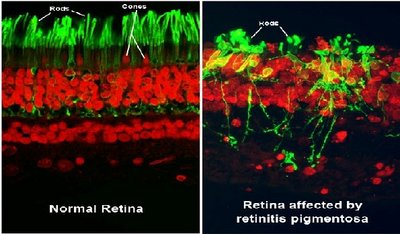
Las distrofias de conos y bastones (CRDs) son distrofias hereditarias de la retina. Se incluyen en el grupo de la retinosis pigmentaria, y, de forma más general, en el de las retinopatías pigmentarias.
Como tal, se caracterizan por la presencia de depósitos de pigmentos en la retina, visibles en la exploración del fundus, que se localizan predominantemente en la región de la mácula. A diferencia de las retinosis pigmentarias típicas (también conocidas como distrofias de bastón-cono, RCDs), que son el resultado de una pérdida primaria de función de los fotoreceptores de los bastones, seguida por una pérdida secundaria de la función de los fotorreceptores de los conos; las distrofias de conos y bastones reflejan una secuencia de hechos totalmente opuesta, con una implicación primaria de los conos, o con una pérdida de función concomitante de conos y bastones.
Esto explica la naturaleza de los síntomas de las distrofias de conos y bastones, agudeza visual disminuida, defectos en la visión de los colores, fotoaversión y disminución de la sensibilidad en el centro del campo visual, seguido por una pérdida de la visión periférica y ceguera nocturna.
Por tanto, el curso clínico de las distrofias de conos y bastones suele ser más grave y rápido que el de las distrofias de bastón-cono. Provocan una ceguera legal más temprana (agudeza visual inferior a 20/200) e invalidez. En la etapa final, sin embargo, las distrofias de conos y bastones no son diferentes de las distrofias de bastón-cono.
¿Son sindrónicas?
Las distrofias de conos y bastones no suelen ser sindrómicas, aunque pueden formar parte de diversos síndromes, tales como el de Bardet-Bield y la ataxia cerebelar SCA7.
Prevalencia y genes implicados
Las distrofias de conos y bastones tienen una prevalencia estimada de 1/40000. Las distrofias de conos y bastones no sindrómicas son genéticamente heterogéneas, con 10 genes clonados y tres loci identificados.
Sigue sin estar claro el porcentaje de pacientes para el que los genes clonados aportan una explicación. De todas maneras, entre los genes ya clonados se han identificado cuatro genes principales para las distrofias de conos y bastones:
- ABCA4, que provoca la enfermedad de Stargardt, además del 30 a 60% de las distrofias de conos y bastones autosómicas recesivas
- CRX y GUCY2D, que son responsables de muchos casos de distrofias de conos y bastones autosómicas dominantes
- RPGR, que provoca cerca de 2 tercios de las RPs ligadas al cromosoma X, además de un porcentaje no determinado de las distrofias de conos y bastones ligadas al X.
Por tanto, hay una gran heterogeneidad en los genes responsables y los mecanismos implicados en las distrofias de conos y bastones. De todas maneras, es probable que haya otras mutaciones muy deletereas en genes, que de otra forma conducirían a RP o a distrofia macular, puedan provocar distrofias de conos y bastones.
¿Cómo se diagnostica?
El diagnóstico de las distrofias de conos y bastones se basa en la historia clínica, el examen del fundus y en los resultados del electroretionograma, que suelen presentar una mayor disminución inicial de la amplitud de las respuestas fototópicas que de las respuestas escotópicas, antes de pasar a ser indetectables. Las complicaciones y el tratamiento para este trastorno son los mismos que para las RPs.